|
|||||||||
|
|
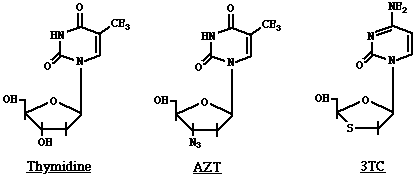
Lamivudina (3TC), Didanosina(ddI), Zalcitabina (ddC), Stavudina (d4T): Tienen mecanismos de acción similares al AZt. 3TC = 2'-deoxy-3'-thiacytidine; ddI: 2',3'-dideoxyinosina; ddC: 2',3'-dideoxycytidina; d4T: 2',3'-didehydro-3'-deoxythymidina. Estos son análogos dideoxynucleosidos del dC, dA, dC, y dT respectivamente.
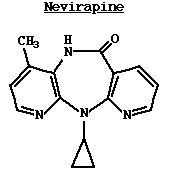
Nevirapine: Es un inhibidor no nucleósido de la trancriptasa inversa (RT). A diferencia de los análogos de los nucleótidos, para ser activa no necesita de una fosforilación, su acción es unirse a la RT e inactivar el enzima de una manera no competitiva. La nevirapina se une directamente a un hueco de 3 dimensiones cerca del sitio activo dentro de la molécula del enzima de la RT. Inhibidores de la integrasa
Una de las más nuevas clases de drogas bajo desarrollo que parece ser alentadora es el inhibidor de integrasa. La integrasa es una enzima usada por el VIH para penetrar su DNA en las células CD4 T. Los inhibidores de integrasa han estado bajo estudio por años, pero hasta ahora han tenido muchos efectos secundarios para tomar un paso adelante hacia ensayos clínicos que envuelvan a participantes humanos. Shiongi, una compañía japonesa, reportó a su nuevo inhibidor de integrasa llamado S-1360. S-1360 está en sus primeras etapas de desarrollo, sin embargo, ha demostrado una buena actividad en contra del virus resistente a todas las clases terapéuticas disponibles actualmente, en tubos de ensayo y pocos efectos secundarios en pruebas con animales. Desde la conferencia Shiongi ha unido sus fuerzas con GSK para formar la farmacéutica Shionogi-GlaxoSmithKline e iniciar la Fase I de los ensayos clínicos con S-1360. En una reunión reciente de GSK se discutieron los planes de las Fases I/II para ensayos en los Estados Unidos. Ningún inhibidor de la integrasa ha sido todavía aprobado su mecanismo de acción radica en el hecho de impedir que el ADN viral se introduzca en el ADN de la célula. Inhibidores de proteasa
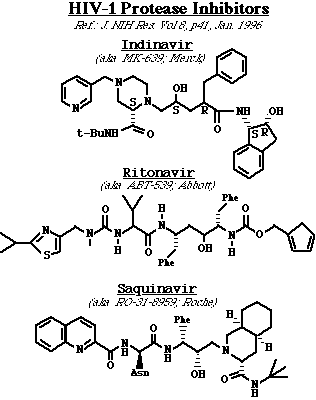
Inhibidores de la proteasa del VHI. La VHI PR es un dímero simétrico, que contiene residuos apareados de ácido aspártico en su sitio activo. El complejo enzima-substrato también contiene una molécula de agua muy importante, única en las proteasas retrovirales, comparada con otras proteasas de ácido aspártico. La mayoría de los inhibidores enzimáticos son compuestos que imitan a los péptidos que son análogos de estados de transición. En otras palabras, dichos compuestos imitan la configuración del estado transicional del substrato natural del VHI PR. Como consecuencia, los inhibidores se unen al enzima más fuertemente que el substrato natural (ya que el substrato debe distorsionarse para asumir la configuración de estado de transición). Estos compuestos son por lo tanto, inhibidores enzimáticos competitivos, y se clasifican de acuerdo a la simetría de sus estructuras. Así, el inhibidor de Merck, Indinavir y el inhibidor de Roche, saquinavir son asimétricos, mientras que el ritonavir de Abbot es muy simétrico. En general, los inhibidores de la proteasa han mostrado una gran promesa como fármacos anti VHI, particularmente cuando se usan en combinación con otros fármacos anti-retrovirales. Los problemas presentados con los inhibidores de la proteasa incluyen (1):mala absorción por vía oral (solo una pequeña fracción ingerida, pasa al torrente circulatorio), y (2): cruzan muy mal la barrera hemato-encefálica y por ende muy bajas concentraciones en el tejido cerebral.
Terapia combinada. La terapia combinada ó el uso simultáneo de múltiples fármacos antivirales anti-VHI, puede ser el medio más efectivo de controlar la infección por VHI (por ejemplo: AZT + 3TC + indinavir es una combinación efectiva, tanto como otras combinaciones). Nota importante: en la clínica diaria se han descrito mutantes del VHI-1 resistentes a todos los fármacos descritos en esta hoja. La resistencia viral es un problema muy importante en los tratamientos antivirales. Los inhibidores de proteasa modificaron de manera radical la historia natural de la infección por VIH, especialmente en los pacientes en estados avanzados. Gracias a ellos, la mayoría de los sujetos en etapas terminales son capaces de recuperar inmunidad, peso corporal, disminuir o desaparecer síntomas sistémicos y mejorar notoriamente su calidad de vida de una manera sostenida. Si bien es cierto que estos mismos efectos se podían observar con la administración de uno o dos análogos de nucleósido, esto se lograba sólo en un 10% de los pacientes, y al cabo de los meses los efectos benéficos iban desapareciendo. La propiedad que marca esta diferencia es la potencia elevada antirreplicativa de la mayoría de los inhibidores de proteasa. Este grupo de medicamentos inhibidores de VIH actúan inhibiendo, es decir evitando el funcionamiento, de una proteína funcional del VIH con actividad enzimática de proteasa sobre otras moléculas estructurales del propio virus. Impiden la maduración de los nuevos virus, impidiendo que se vuelvan infecciosos los nuevos virus producidos. Entonces los inhibidores de proteasa impiden nuevos ciclos de infección a otras células, pero no actúan sobre la replicación viral en células ya infectadas, como si lo hacen los inhibidores de transcriptasa reversa nucleósidos y no nucleósidos. RESISTENCIA A INHIBIDORES DE PROTEASA La resistencia surge del efecto inhibitorio parcial de un inhibidor de proteasa debido a una dosis menor a la recomendada, pobre absorción, por ejemplo el invirase, ingesta irregular o interacción medicamentosa desfavorable, como el indinavir en dosis usual más efavirenz. El mecanismo molecular es la aparición de mutación en los codones de la enzima proteasa o en la poliproteina Gag - Pol que disminuyen la afinidad de la proteasa por el inhibidor de proteasa. Todos los inhibidores de proteasa gasta ahora en el mercado pueden desarrollar resistencia. Existen mutaciones primarias y secundarias, mutaciones en el sitio activo y mutaciones fuera del sitio activo. La mutación primaria es aquella que confieren una resistencia considerable al inhibidor de proteasa; por el contrario, las secundarias complican la terapéutica pues amplían el espectro de resistencia a varios inhibidores de proteasa. Las pruebas moleculares de resistencia como ensayos genotipicos y fenotipicos, son de interpretación difícil y actualmente tienen una utilidad limitada en la aplicación clínica. NUEVOS INHIBIDORES DE PROTEASA
Hay inhibidores de proteasa en diferentes fases de estudio que muestran ventajas en relación a los primeros cuatro. Destacan amprenavir: VX478, agenerase AG-.1778, AB-378, tripanavir: PNU-140690, BMS 232632 y AG-1776. La mayoría de ellos tienen un bajo nivel de resistencia cruzada con los primeros cuatro inhibidores de proteasa pero sólo los nuevos estudios controlados determinaran el verdadero peso de estos nuevos medicamentos en segunda opción de inhibidores de proteasa. Hay ventajas potenciales adicionales como el compuesto BMS 232632 que administrado una vez al día es capaz de alcanzar niveles sanguíneos inhibitorios adecuados. |
|
|