|
|||||||||
|
|
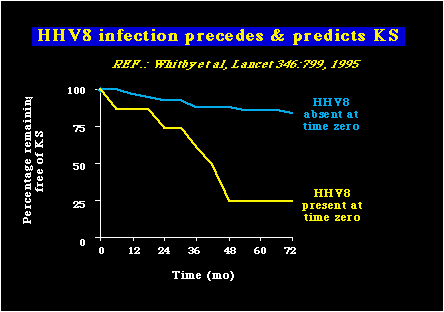
El sarcoma de Kaposi asociado al Herpesvirus (KSHV) también llamados: Human herpesvirus 8 (HHV-8)Herpesvirus Humano 8 (HHV-8) : Fué descubierto por Yuang Chang y Patrick Moore en 1994, durante los estudios que hicieron para estudiar la etiología de un importante tumor asociado al SIDA, conocido como el Sarcoma de Kaposi (KS). Por algunos años se pensó que el KS podría tener un origen infeccioso, debido a que este tumor es frecuente en homosexuales masculinos infectados con HIV y sin embargo es raro en otros grupos de personas infectadas con el HIV. Moore y Chang usaron un método novísimo conocido como Análisis de Diferencias Representativas (RDA=Representational Difference Analysis), para comparar muestras de ADN preparadas de tumores del Sarcoma de Kaposi con muestras de ADN de personas sin el Sarcoma de Kaposi. Durante éstos análisis, ellos encontraron secuencias de ADN que sólo estaban presentes en los tejidos KS. Resultaron ser fragmentos del genoma del HHV-8. Muchos estudios han aportado evidencia convincente de la asociación del HHV-8 con el KS. Uno de los estudios fué dirigido por Robin Weiss y sus colegas, quienes usaron la PCR para detectar el ADN del HHV-8 en la sangre de las personas infectadas con HIV. Ellos separaron sus enfermos en dos grupos (HHV8+ con tiempo cero y HHV8- con tiempo cero), y entonces se hicieron la pregunta: ¿Cuál es la probabilidad de desarrollo de KS en estos dos grupos? Los resultados se muestran a continuación.
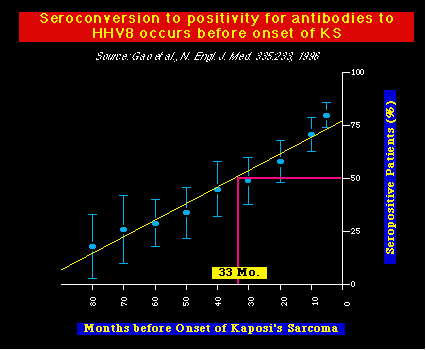
Evidencia adicional de una asociación causal entre el HHV8 y el KS fué dada por Chang y Moore, quienes usaron un parámetro diferente para evaluar la infección por HHV8. En este caso, ellos examinaron un grupo de individuos infectados con HIV para detectar la presencia de anticuerpos en contra del HHV8. Después, ellos midieron el tiempo que se necesitaba para el desarrollo del KS. En promedio, hallaron que la seroconversión a la positividad para el HHV8, predecía y precedía la aparición del KS, en aproximadamente 3 años.
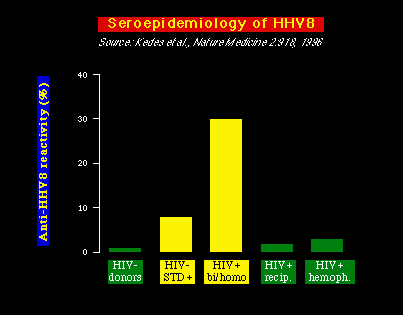
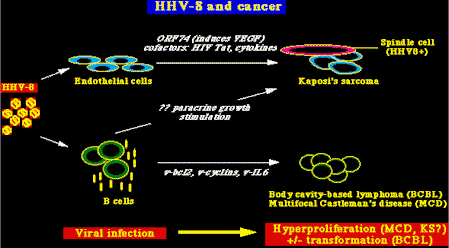
También se ha encontrado que el HHV8 está asociado con ciertos tumores de células B en personas infectadas con HIV, conocidos como linfomas de efusión primarios (sinónimo: linfomas de células B de las cavidades corporales ). Además, se ha demostrado que el virus infecta preferentemente las células B. El HHV8 se ha encontrado en tejido prostático y en el semen humano, sugiriendo un posible mecanismo de transmisión viral. Hay datos adicionales que apoyan la idea de que el HHV8 se transmite sexualmente. Primero: Dean Kedes y sus colegas escrutinizaron muestras de sangre para detectar la presencia de anticuerpos contra el HHV8 y encontraron que los anticuerpos contra el HHV8 son relativamente frecuentes en personas HIV negativas que consultaban en clínicas de Enfermedades de Transmisión Sexual (ETS), personas HIV-ETS+, comparadas con donantes de sangre HIV- (Donantes HIV-). Y también, y consistente con la epidemiología de la enfermedad, HHV8 es común en hombres homosexuales y bisexuales HIV+ (quienes comúnmente desarrollan KS), pero es mucho más raro en otros grupos de personas HIV+, como los hemofílicos (hemoph HIV+) ó recipientes de transfusiones de sangre (recip HIV+), grupos que rara vez desarrollan el KS
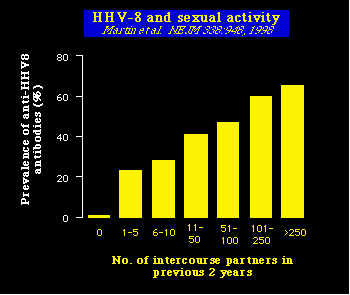
Segundo, este mismo grupo de investigadores también encontraron que el riesgo de infectarse con el HHV8 (definido por la presencia de anticuerpos anti HHV8) es directamente proporcional al número de personas con las que se ha tenido relaciones sexuales.
La noción de que el HHV8 es un virus de transmisión sexual puede explicar por qué éste virus, en contraste con la mayoría de los demás herpesvirus, es relativamente raro en la mayoría de la gente. En este sentido, es instructivo considerar el HSV2, el cual también se transmite sexualmente y es mucho menos frecuente que los demás herpesvirus, desde el 1 al 7. Genoma del HHV8. Al final de
1966, las secuencias de todo el genoma del HHV8 fué estudiado por varios
investigadores. Se encontró que el genoma viral contenía algunos genes muy interesantes,
incluyendo homólogos de varios genes celulares (Moore et al. Science 274:1739, 1996),
entre ellos:
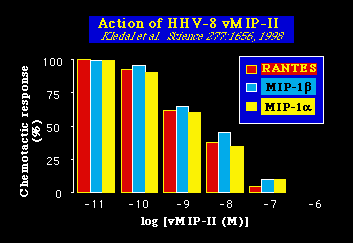
El homólogo de la quimiocina viral, v-MIP-II, tiene la rara propiedad de poder inhibir la acción normal de un amplio espectro de quimiocinas celulares. Dichas quimiocinas (entre ellas RANTES, MIP-1a y MIP-1b; ver más abajo) habitualmente ayudan a reclutar células inflamatorias a los sitios de infección por quimiotaxis. El amplio espectro de actividad, un hecho raro, del v-MIP-II, lo hace interesante como un compuesto de base, del cual poder desarrollar nuevos fármacos anti HIV, que puedan bloquear la interacción entre HIV y los múltiples receptores de las quimiocinas.
No está claro cuál, si es que hay alguno, de los productos génicos del HHV8 puede ser importante en el desarrollo del KS y de otros tumores asociados al HHV8. (Ver más abajo). Es evidente que la patogenia del KS no está bien entendida y que posiblemente sea compleja (Gallo, Science 282:1837, 1998). Características principales del KS, entre otras:
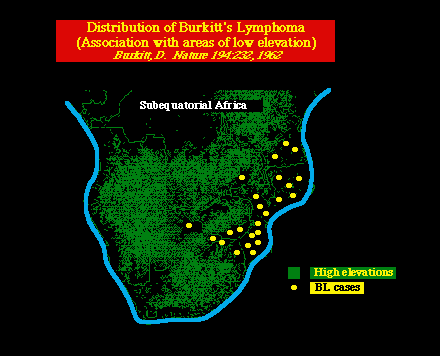
Virus de Epstein BarrRef.: Sugden, Sem. Virol. 5:197-205, 1994 Descubrimiento del EBV. El EBV fué descubierto por Denis Burkitt en 1950. El identificó una forma de cáncer, previamente pasada por alto, que afectaba la mandíbula de los niños y adolescentes africanos, y él tuvo la crucial inspiración de que la distribución de este tumor tan común, parecía estar influenciado por factores climáticos, notablemente la temperatura y la altitud. Burkitt teorizó, que el tumor podía ser debido a un virus transmitido por mosquitos ó a un arbovirus. Distribución geográfica del linfoma de Burkitt. Los hallazgos están asociados con zonas de poca elevación.
Este descubrimiento condujo a Tony Epstein, Yvonne Barr y Burt Achong a examinar las biopsias de tumores recién extirpados, buscando un virus. En 1964, con un microscopio electrónico, encontraron un virus parecido a las partículas de los herpesvirus, en un pequeño número de células biopsiadas, y así establecieron que de hecho éste era un virus nuevo. El virus de Epstein-Barr fué así nominado como el primer candidato causal de un tumor humano. El EBV es un miembro de los linfocriptovirus, género de los gammaherpesvirus. Virus relacionados existen en los primates del Viejo Mundo, incluyendo los Pan Herpesvirus y Papio Herpesvirus . Estos virus comparten un tropismo hacia los linfocitos B, y tienen una tendencia a la oncogenicidad. Propiedades biológicas del EBV: generalidades: Como los demás herpesvirus, EBV infectan las células que no se dividen. Pero, en el caso del EBV, las células que infecta son principalmente linfocitos B primarios, y produce una infección latente con una excelente eficiencia, tanto in vitro como in vivo. El virus induce la proliferación de dichas células y sostiene la multiplicación por medio de la expresión de genes específicos para el crecimiento celular, con la habilidad de convertir al EBV en tumorogénico. Las células B infectadas de modo latente expresan solo el 10% del genoma del EBV y así rara vez apoyan la fase lítica del ciclo vital del EBV. La infección latente de las células B por el EBV representa el ejemplo mejor entendido de la latencia de los herpesvirus. Esta charla, se centra exclusivamente en la fase latente de la infección por EBV. Infección latente de las células B por el EBV. Los pasos en la infección de las células B son:
La habilidad extraordinaria del EBV para inducir eficientemente una transformación del crecimiento de las células B en un permanente estado de líneas celulares linfoblásticas, es un proceso que tiene varias etapas.
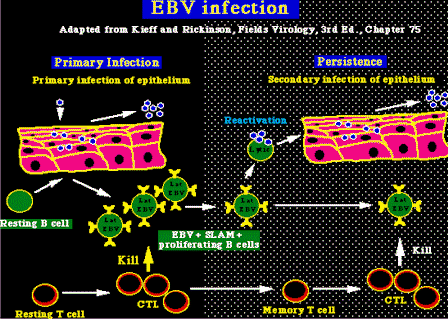
Ciclo lítico del EBV. In vivo, la replicación del EBV se hace más eficientemente en las células epiteliales, donde se cree que entra el virus con la ayuda de la molécula CD21 pero la replicación del EBV, también ocurre espontáneamente en una pequeña fracción de la población de las células B infectadas de forma latente, como resultado de la reactivación viral. No se conocen los estímulos para este fenómeno, a pesar de que la replicación de una pequeña parte del EBV en las células B infectadas puede estimularse in vitro, usando un éster del phorbol, (un activador de la proteín-cinasa C ) ó por la expresión de un poderoso trans-activador inmediato temprano de la fase lítica de la infección por EBV: la proteína codificada BZLF1. Patogenia del EBVGeneralidades: Las enfermedades asociadas con el EBV aparecen, corrientemente, por un fallo en la respuesta inmune del huésped en el control de la proliferación de las células infectadas de modo latente. Es lo opuesto de lo que pasa con los otros herpesvirus, donde el problema es un fallo en la respuesta a la infección lítica, y esto se correlaciona con el hecho de que las infecciones latentes por el EBV, predominan in vivo, con la fase lítica de la infección afectando a muy pocas células. Patogenésis de la infección por EBV. La infección es por lo general asintomática en la niñez, y casi el 90% de los adultos son positivos al EBV. Se propaga por la saliva y el virus infecta al principio las células epiteliales de la orofaringe, donde se replica muy bien. Después el virus infecta las células T, a medida que circulan por la orofaringe y acaba con el establecimiento de una infección latente de las células B. El EBV persiste durante toda la vida en las células B, con aproximadamente 1 en 105 a 106 células B positivas al virus. La infección de las células B está asociada con la veloz proliferación y expansión de las células B EBV+ en la fase primaria de la infección viral. Normalmente, ésta multiplicación celular dirigida por el EBV queda después bajo el control de las células citotóxicas T (CTLs). Lo que da lugar a la mononucleosis infecciosa (MI), usualmente en los adultos jóvenes. Pero, en ciertas personas, la proliferación celular impulsada por el EBV, no es controlada apropiadamente, y puede dar lugar a una MI fatal, hecho que ocurre en especial, en varones con una alteración linfoproliferativa ligada al cromosoma X (XLP) Papel del SLAM y del SAP en la patogenia de la infección por EBV. SLAM (Signalling Lymphocyte-Activation Molecule = molécula de señalización de activación del linfocito), es una proteína de la superficie celular de las células B y T y tiene una alta afinidad para unirse a si misma, capaz de co-activar las respuestas de las células B y T, en combinación con los receptores de las células B y T. Las células B transformadas por el EBV, expresan SLAM con niveles altos y son células funcionantes presentadoras de antígeno.(por ejemplo tienen la facultad de estimular la proliferación de las células B). Así, se piensa que SLAM es importante para la generación de respuestas específicas de las células T del EBV, el cual podría controlar la proliferación de las células infectadas B. Se ha demostrado recientemente que el gen XLP es responsable de codificar una proteína conocida como SAP (Slam Associated Protein = Proteína Asociada al SLAM). SAP actúa como un inhibidor natural de SLAM, y mutaciones en el gen SAP en los pacientes con XLP, llevan a una ausencia funcional de la proteína SAP. Esto a su vez afecta las interacciones celulares T/B iniciadas por SLAM, resultando en la imposibilidad de controlar la proliferación celular B inducida por el EBV.(Consulte Klein, Nature 395:441, 1998; Sayos, Nature 395:462, 1998; Coffey, Nature Genetics 20:129, 1998)
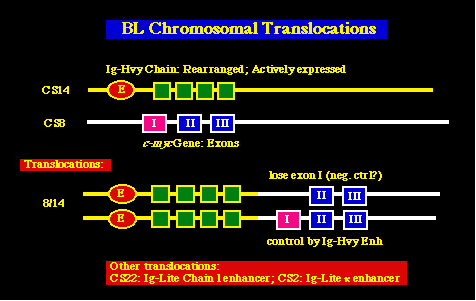
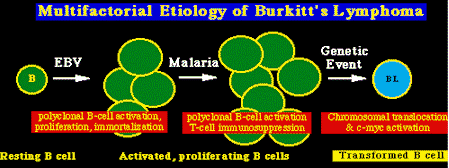
Tumores asociados al EBVLinfomas inmunoblásticos. La infección latente por EBV de las células B está asociada con un desarrollo rápido del tumor en personas inmunocomprometidas, como son los recipientes de transplante de médula ósea y personas con SIDA (10% formarán tumores de células B) Linfoma de Burkitt. El linfoma de Burkitt (BL) también está relacionado con la inmunosupresión en sujetos con SIDA, y a la malaria en los casos de endemia de BL. Este último fenómeno explica los hallazgos originales de Burkitt, donde los factores climáticos están ligados a la etiología de la endemia de BL, ya que la temperatura y la altitud condicionan el crecimiento y distribución de los mosquitos que transmiten la malaria. Todos los casos de BL están también marcados por la presencia de traslocaciones cromosómicas específicas, las cuales producen la activación del c-myc proto-oncogene. La mayoría de éstos comprenden una traslocación recíproca entre el cromosoma 8, en , ó cerca del c-myc loci, y el loci de las cadenas pesadas de las inmunoglobulinas en el cromosoma 14.
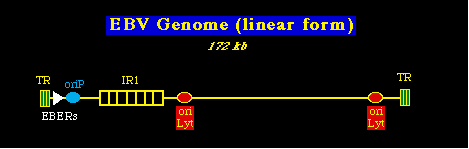
Otros tumores asociados al EBV incluyen la enfermedad de Hodgkin (linfoma maligno), ciertos tipos raros de linfomas de células T, y un tumor epitelial que se desarrolla después de muchos años de la infección inicial y es común en el Sureste de Asia: el carcinoma nasofaríngeo. Tabla con tumores asociados al EBV Biología Molecular y el GenomaGenoma del EBV. Fué el primer herpesvirus cuya secuencia se estudió completamente en 1984. Tiene un tamaño de 172 kbp y contiene una sola región larga que está interpolada con varios repetidos internos y flanqueada por repetidos directos terminales en cualquiera de sus terminaciones. El más grande de los repetidos internos se conoce como IR1 y corresponde al repetido Gly-Ala que es partte de la proteína EBNA1. NOTA: han sido identificados dos tipos distintos de EBV: EB1 y EB2, los cuales tienen una gran parecido y son igualmente frecuentes en diferentes habitats. El significado de éstos diferentes tipos de virus está por determinar.
Genes inmortalizadores del EBV. Se ha demostrado que son necesarios 4 genes latentes del EBV para que se efectúe una proliferación de células infectadas cuantificable. Son: EBNA2, LMP1, EBNA3A y EBNA3C. Una quinta proteína, EBNA1, también es necesaria para el mantenimiento del genoma EBV en las células infectadas de modo latente. Estos genes inmortalizadores del EBV tienen 3 importantes características:
Tabla que sumariza los genes inmortalizadores del EBV Patrones de expresión de los genes latentes del EBV. Hay varios: Uno está involucrado en el mantenimiento del genoma en la fase de reposo de las células B. Este hecho puede ocurrir en ausencia de la expresion EBNA1, ya que no hay necesidad para el EBV de replicar su genoma en una célula B en reposo. Lo que no está claro, es cómo una célula B infectada con EBV puede volver al estado de reposo. Otros genes son:
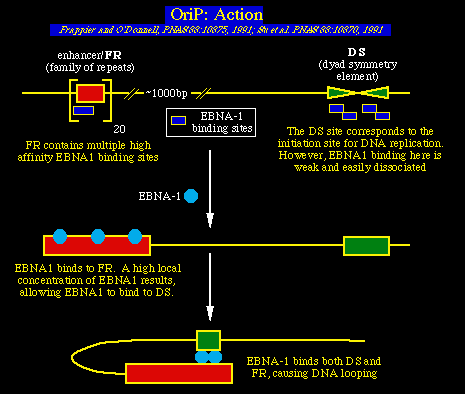
OriP y la fase latente de la replicación ADN del EBVOri es el origen de la síntesis de ADN para el episoma del ADN del EBV en las células B infectadas de modo latente. Su actividad depende totalmente de la expresión de EBNA1, ya que también permite la replicación del plásmido en las células de los primates, células que expresan solamente el EBNA1 y no los demás genes del EBV. La actividad OriP posiblemente requiera también una ó más proteínas celulares, ya que el OriP no permite la replicación del plásmido en las células que no son de los primates y que sin embargo expresan EBNA1. Más experimentos que resultaron en la identificación original de oriP. OriP está compuesto de dos elementos. Uno es el sitio FR (una familia de repeticiones) y el otro es el elemento DS (simetría dyad), el cual está localizado a 1 kb de distancia. La replicación del ADN empieza en el elemento DS, lo que es sorprendente, pues el elemento DS se une al EBNA1 mucho más débilmente que el elemento FR. Pero los estudios de Lori Frappier, Bill Sugden y sus colegas revelaron que el FR actúa como un intensificador de la replicación.. Específicamente, el FR se une al EBNA1 de modo eficiente. El resultado es que EBNA1 se acumula en el sitio del genoma EBV (FR) muy cercano al sitio DS. Este hecho facilita el contacto de EBNA1 con el elemento DS y a la vez interactúa con el elemento FR (lo que se explica porque el EBNA1 puede unirse a sí mismo como también al ADN).
Mas información acerca del trabajo de Edwards, ver la estructura cristalina del EBNA1, cuando se une al ADN.
Actualizado el 1/99, Steve Dewhurst, Associate Professor of Microbiology
and Immunology URL: http://www.urmc.rochester.edu/smd/mbi/grad2/herp99B.html
|
|
|